Emily Huynh & Shannon Chau

")


Overview of Lyme Disease and the Lyme Project
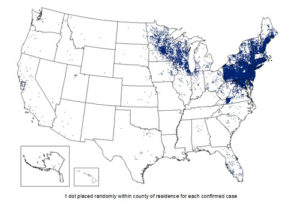
Lyme disease is a multi-organ illness that is transmitted to humans via ticks. Ticks act as a vector for the disease-causing spirochete Borrelia burgdoferi. Lyme disease was traditionally prevalent in the Northeast region, but is now found in all fifty states. Cases have risen to over 300,000 each year (Center for Infectious Disease Control and Prevention). To this end, Lyme disease is now the most common vector-borne illness, and it ranks number six in most commonly reported infectious diseases. This rapid increase in Lyme disease cases poses a concern for both researchers and public health overall.
Signs and symptoms of Lyme disease include a signature eyrthem a migrans rash (a red, bullseye-shaped mark), fever, chills, and headache. Oral antibiotics taken in early states of acute Lyme disease usually help patients recover fully. However, chronic lyme disease, also known as Post-Treatment Lyme Disease Syndrome (PTLDS), can occur. This is a condition in which the patient has symptoms long after treatment has ended. The causes and long-term effects of PTLDS are still unknown. In addition, diagnosis of PTLDS is surrounded by controversy due to a lack of consensus and misconceptions within the medical community.
Thus, to address the rapid spread of Lyme disease and deficient knowledge surrounding its chronic form, researchers at ISB are studying the condition through many approaches including proteomic analysis.
Proteomic Training/Skills
In order to be able to execute the lab work necessary for their project, Emily and Shannon spent a portion of the summer adding to their lab skill set. They learned BCA and Micro-BCA Protein Assays used for protein quantification. These techniques are relatively quick and easy for providing more information on the sample at hand.

An example of one of the BCA protein assays that provided information for the S-Trap protocol.
Another useful method that Emily and Shannon learned was gel electrophoresis. Initially, the two were taught how to run a gel for DNA. As their project developed, they learned how to apply the same principles to running a gel for proteins. This was accompanied with learning how to view the protein bands through silver staining.
As a result of their constant work with protein samples, Emily and Shannon had to quickly learn the proper way of handling proteins. In order to avoid protein degradation, proteins had to be properly stored at -80°C for long periods of time, -20°C for short periods of time, and on wet or dry ice while executing work.
S-Trap Protocol (Main Focus)
Optimization of Sample Preparation in Proteomic Analyses Using Suspension Trap Digestion
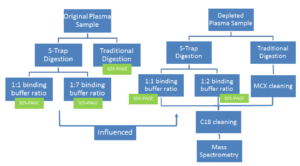
One of the essential parts of studying proteomics revolves around sample preparation, which is used to digest proteins down to their peptides. Research scientists can spend significant amount time and money preparing hundreds of samples at a time. One of the current ways that ISB prepares their protein samples for mass spectrometry is referred to as “traditional digestion.” Traditional digestion is an in-solution digestion that involves a variety of steps — most notably, an overnight incubation period that can hinder lab productivity. To streamline the process, Emily and Shannon adjusted and evaluated a more efficient protocol using the suspension trap method, based on the S-Trap ™ column (a product from the biotechnology company, ProtiFi, LLC). The protocol that they created can be used to expedite sample preparation of Lyme disease patients’ plasma samples.
Lyme disease patient samples are often processed as depleted plasma. Unlike tissue samples (for which the original protocol was designed), depleted plasma samples lack cellular material that needs to be broken down. In addition, depleted samples have a much higher volume and lower protein concentration than pure plasma.
Therefore, they considered the following obstacles when adjusting the protocol:
- Side effects due to overuse of strong chemicals for digestion
- The low protein concentration
- Incomplete protein digestion/protein loss
To account for these factors, Emily and Shannon tested multiple areas for revision. For one, they reduced reagent concentrations to avoid over use of the chemicals. They also tested multiple binding buffer ratios to evaluate how little they could add without compromising protein retention.
Later, Shannon and Emily utilized the proteomic skills of SDS-PAGE gel electrophoresis and silver staining to qualitatively analyze the S-Trap column’s protein-binding power–and therefore its ability to decrease protein loss in comparison to traditional digestion. They ran gels on the flowthrough from the binding, washing, and trypsin digestion steps of the protocol, as well as the final elution and the traditional digestion. Then they noted how many proteins would have been lost as flowthrough in each step (had the flowthroughs been disposed) by comparing them to the final S-Trap eluted proteins and the traditional digestion proteins.


The SDS-PAGE on the left was used as an initial, qualitative evaluation method for the S-Trap. This gel has “blank” lanes in lanes x-x, which showed that protein loss was minimal in all of the flowthrough steps except for the addition of digestion buffer (labeled “Trypsin 1:1” and “Trypsin 1:7” in the gel lanes). Knowing this, Emily and Shannon repeated the experiment with a few adjustments.
First, the higher binding buffer to sample volume ratio was decreased to a 1:2, rather than 1:7, since the 1:7 setup did not lose significant amounts of peptides. Second, all flowthrough was disposed of except for that of the binding step (named “Binding FT”) because the experimenters wanted to continue testing different binding buffer ratios. Finally, Shannon and Emily decided to use a shorter centrifugation for the digestion buffer and to return all digestion buffer flowthrough to the top of the column. This was in attempt to reduce the peptides lost in the digestion buffer step prior to incubation. All of these changes proved to be successful once they were loaded into the second gel on the right.
From there, the second trial’s design was repeated on depleted plasma samples (rather than original plasma samples) that were loaded into a mass spectrometer for a quantitative analysis of peptide retention.
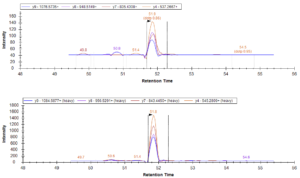

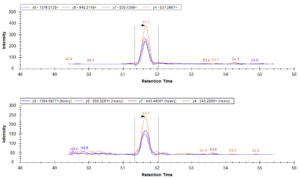
Conclusions:
Data from the mass spectrometer led Shannon and Emily to the following conclusions:
- Based on the data collected, the S-Trap provides a sufficient alternative to in-solution digestion: retained peptides were at similar or higher levels than the traditional method. In particular, the S-Trap method with a binding buffer to sample ratio of 1:1 had the best peptide retention in this trial.
- The S-Trap protocol can be applied to larger volume depleted plasma samples without interfering with efficiency and accuracy.
- Further experimentation (repeated trials) will be necessary to confirm these results, but the S-Trap has strong potential and viability for use with ISB’s Lyme Disease research.
For more information:
On the S-Trap project: Click here
On the ISB Lyme project: Click here
On Lyme disease: Click here
Contact us: e.huynh247@gmail.com, shannonchauusa@yahoo.com