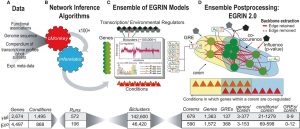
 We previously constructed an “Environment and Gene Regulatory Influence Network” (EGRIN) for Halobacterium salinarum NRC‐1 (Bonneau et al, 2007). This model was constructed in two steps. First, modular organization of gene regulation was deciphered through semi‐supervised biclustering of gene expression, guided by biologically informative priors and de novo cis‐regulatory GRE detection for module assignment (cMonkey; Reiss et al, 2006). Second, using a regression‐based approach, transcriptional changes of genes within each bicluster were modeled as a linear combination of influences of TFs and environmental factors (Inferelator; Bonneau et al, 2006). While full description of these algorithms is beyond the scope of this work, readers are encouraged to refer to the original papers and Supplementary Information for more detail.
We previously constructed an “Environment and Gene Regulatory Influence Network” (EGRIN) for Halobacterium salinarum NRC‐1 (Bonneau et al, 2007). This model was constructed in two steps. First, modular organization of gene regulation was deciphered through semi‐supervised biclustering of gene expression, guided by biologically informative priors and de novo cis‐regulatory GRE detection for module assignment (cMonkey; Reiss et al, 2006). Second, using a regression‐based approach, transcriptional changes of genes within each bicluster were modeled as a linear combination of influences of TFs and environmental factors (Inferelator; Bonneau et al, 2006). While full description of these algorithms is beyond the scope of this work, readers are encouraged to refer to the original papers and Supplementary Information for more detail.
The EGRIN networks learned by cMonkey and Inferelator accurately predicted transcriptional changes in new environments, a feat that has subsequently been replicated by other network inference strategies (Faith et al, 2007; Lemmens et al, 2009; Marbach et al, 2012); yet, these network models have failed to capture detailed regulatory mechanisms that operate only in specific environments, at non‐canonical genomic locations, or in complex combinatorial schemes.
We made significant advancement to inference of GRNs that overcomes many of these challenges. We have developed a methodology applicable to any sequenced microbe in culture to infer EGRIN 2.0 models for two representative organisms from the primary branches of prokaryotic life—bacteria and archaea: (1) Escherichia coli, a bacterium with a wealth of information about transcriptional regulatory mechanisms and related experimental data (Salgado et al, 2012); and (2) H. salinarum, an archaeon with few examples of regulatory mechanisms that have been characterized in detail, but extensive experimental data from recently conducted systems biology studies (Bonneau et al, 2007; Koide et al, 2009). The wide range of prior knowledge for these organisms proved invaluable for testing our model. In addition, we have also conducted new experiments that validate EGRIN 2.0‐predicted complex modulation of the E. coli transcriptome structure during varying stages of growth in rich media.
EGRIN 2.0 models the organization of GREs within every promoter and their distributions across the entire genome—even in non‐canonical locations—and links the contexts in which they act to conditional co‐regulation of genes. These features are formalized in EGRIN 2.0 by condition‐specific, co‐regulated modules or corems. Corems are overlapping sets of co‐regulated genes that, in some cases, group together genes from different regulons and, in other cases, subdivide genes of the same regulon, or even the same operon. EGRIN 2.0 formalizes how the genome‐wide coordination of previously characterized and newly discovered regulatory mechanisms dynamically associates genes into corems, bringing together functionally related genes from different operons and regulons whose deletions have similar impact on cellular fitness. Our results show how prokaryotes, much like eukaryotes, can produce complex gene expression patterns with a relatively small number of regulatory components.