EGRIN
EGRIN (Environment and Gene Regulatory Influence Network).
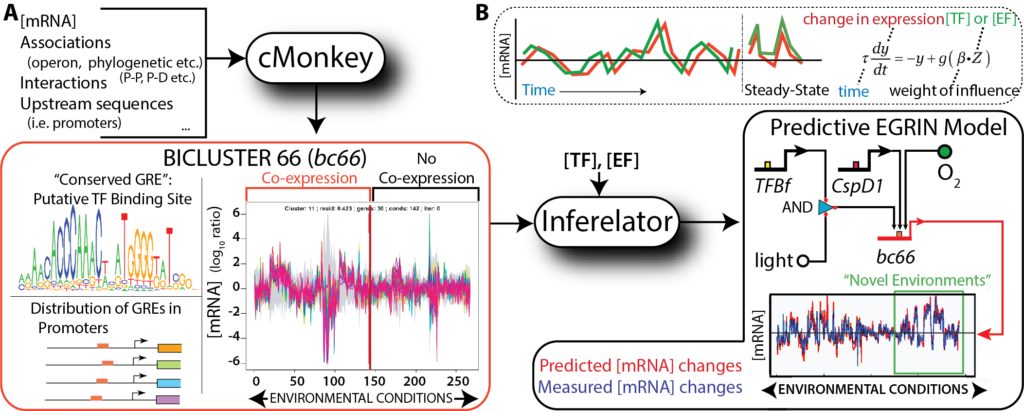
We have developed an approach for data-driven inference of a systems-scale predictive GRN model for any organism that can be cultured in the laboratory. The computational framework is comprised of two algorithms: cMonkey for data integration and biclustering, and Inferelator for inference of regulatory influences. Briefly, cMonkey integrates gene expression, GRE analysis, and gene- gene functional associations to identify groups of conditionally co-regulated genes that are (i) co- expressed in subsets of environments, and (ii) share de novo detected GREs. Inferelator then applies a linear approximation to the dynamical transcriptional rate equations to identify candidate TFs and environmental factors (EFs) that are most likely to activate or repress the genes in each cMonkey-identified module. The resulting EGRIN models cellular regulatory dynamics at multiple scales, from a systems level to specific regulatory mechanisms, and can accurately predict global responses to new environmental and genetic perturbations.
We previously constructed an “Environment and Gene Regulatory Influence Network” (EGRIN) for Halobacterium salinarum NRC‐1 (Bonneau et al, 2007). This model was constructed in two steps. First, modular organization of gene regulation was deciphered through semi‐supervised biclustering of gene expression, guided by biologically informative priors and de novo cis‐regulatory GRE detection for module assignment (cMonkey; Reiss et al, 2006). Second, using a regression‐based approach, transcriptional changes of genes within each bicluster were modeled as a linear combination of influences of TFs and environmental factors (Inferelator; Bonneau et al, 2006). While full description of these algorithms is beyond the scope of this work, readers are encouraged to refer to the original papers and Supplementary Information for more detail.
The EGRIN networks learned by cMonkey and Inferelator accurately predicted transcriptional changes in new environments, a feat that has subsequently been replicated by other network inference strategies (Faith et al, 2007; Lemmens et al, 2009; Marbach et al, 2012); yet, these network models have failed to capture detailed regulatory mechanisms that operate only in specific environments, at non‐canonical genomic locations, or in complex combinatorial schemes.
We have now constructed EGRIN models for more than a dozen organisms. We have shown that EGRIN can effectively predict transcriptional changes in ~80% of all genes in an organism to a novel environmental or genetic perturbation with an average correlation R of ~ 0.8.
We have made further improvements to both cMonkey and Inferelator by integrating them into an ensemble learning approach, which takes advantage of the inherent variability in individual model predictions, to substantially improve the definition of the modular structure of the GRN and the accuracy of predictions EGRIN2) . This updated framework is embarrassingly parallelizable; we have implemented it on Amazon EC2, and have applied for a DOE NERSC ERCAP startup allocation of 1,000,000 hours for significant scale up.
The ensemble-based approach refines the standard definition of a “regulon” by considering the frequency with which regulatory relationships between genes, conditions, and predicted GREs are repeatedly observed - e.g. how often is gene A co-regulated with gene B under condition X, where we have also discovered GRE 1. Post-processing the ensemble allows us to distinguish true signal from noise and to identify environment-dependent nuances in regulation. Using algorithms for detecting modular structures within networks, we can (1) remove noise from the ensemble network, leaving only the statistically significant backbone network62 and (2) group genes into multi-scale conditionally co-regulated modules, or corems, via community detection63. We can then capture the subtle complexities of regulatory relationships between genes, where a small number of genetically distinct regulatory inputs can drive indistinguishable gene activity under some conditions, and we use the term corem to extend the more traditional concept of the regulon into this realm of regulatory combinatorics.