 Mycobacterium tuberculosis (MTB) is an extraordinarily successful pathogen that has infected thirty percent of the world's population. The success of MTB is tied to the adaptive repertoire of the bacilli in the face of varying and hostile environments within the host. In the course of chronic infection, MTB encounters diverse environmental conditions, including hypoxia, nitric oxide stress and varying nutritional limitations. Microbes respond and adapt to such immunological, environmental and nutritional changes through regulatory programs primarily encoded at the transcriptional level. A significant fraction of these regulatory programs are controlled via transcription factors (TFs) that modulate transcriptional activity upon binding to cis-regulatory motifs located in intergenic promoters. A detailed model of MTB's transcriptional regulatory network, including the complete set of TFs, co-regulated genes, and regulatory motifs, has significant implications for elucidating novel strategies to eradicate infection by MTB.
Mycobacterium tuberculosis (MTB) is an extraordinarily successful pathogen that has infected thirty percent of the world's population. The success of MTB is tied to the adaptive repertoire of the bacilli in the face of varying and hostile environments within the host. In the course of chronic infection, MTB encounters diverse environmental conditions, including hypoxia, nitric oxide stress and varying nutritional limitations. Microbes respond and adapt to such immunological, environmental and nutritional changes through regulatory programs primarily encoded at the transcriptional level. A significant fraction of these regulatory programs are controlled via transcription factors (TFs) that modulate transcriptional activity upon binding to cis-regulatory motifs located in intergenic promoters. A detailed model of MTB's transcriptional regulatory network, including the complete set of TFs, co-regulated genes, and regulatory motifs, has significant implications for elucidating novel strategies to eradicate infection by MTB.
Models of transcriptional regulatory networks are typically constructed by integrating large omics datasets with computational algorithms to reproduce and elucidate complex regulatory interactions. Through an iterative process, network models can inform the design of new biological experiments, which yield more powerful models. Bridging the gap between computation and experimentation has remarkable promise, especially in organisms like MTB that are challenging and time-consuming to work with in the laboratory.
Useful information about MTB's transcriptional regulation is available from studies that have focused on regulation during particular stages of pathogenesis such as hypoxia, transition to growth arrest or macrophage infection. However, the aforementioned models cover at most 50% of the MTB genome. To improve on this, we reconstructed a global transcriptional regulatory network model of MTB that encompasses up to 98% of the genome (3922 genes) and accurately predicts gene expression for new environmental conditions.
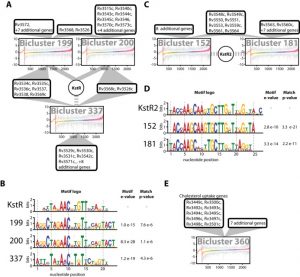
In order to accelerate the discovery and characterization of these adaptive mechanisms, we have mined a compendium of 2325 publicly available transcriptome profiles of MTB to decipher a predictive, systems-scale gene regulatory network model. The resulting modular organization of 98% of all MTB genes within this regulatory network was rigorously tested using two independently generated datasets: a genome-wide map of 7248 DNA-binding locations for 143 transcription factors (TFs) and global transcriptional consequences of overexpressing 206 TFs. This analysis has discovered specific TFs that mediate conditional co-regulation of genes within 240 modules across 14 distinct environmental contexts. In addition to recapitulating previously characterized regulons, we discovered 454 novel mechanisms for gene regulation during stress, cholesterol utilization and dormancy. Significantly, 183 of these mechanisms act uniquely under conditions experienced during the infection cycle to regulate diverse functions including 23 genes that are essential to host-pathogen interactions. These and other insights underscore the power of a rational, model-driven approach to unearth novel MTB biology that operates under some but not all phases of infection.