Integrated systems analysis to characterize adaptation of Mycobacterium tuberculosis to external stimuli
A NOTE FROM THE INTERNS:
Sihan and Jonathan are endlessly thankful for their mentors, Dr. Selva Rupa Christinal Immanuel and Dr. Mario Arrieta-Ortiz for being fantastic mentors with their unwavering support through their learning process, through all of the technical, professional, and interpersonal skills throughout their internship. They would also like to thank the SEE team and everyone whose hard work behind the scenes made their internship possible, especially Claudia Ludwig, Faduma Hussein, Sarah Clemente, Serdar Turkarslan, and Allison Kudla. Huge thanks to the Baliga Lab and everyone at ISB for being so welcoming and making their experience at ISB fun and rewarding. Additionally, they would like to shoutout their fellow interns for staying goofy.





Project Summary
Background and Motivation
Tuberculosis (TB), caused by the bacterium Mycobacterium tuberculosis, remains humanity’s most persistent and deadly infectious disease, responsible for an estimated 1.25 million deaths in 2023.
The success of this pathogen lies in its ability to adapt and survive in the harsh environment of host macrophages within the lung. It withstands hypoxic, acidic, and nutrient-poor conditions, often by entering a dormant state that can persist for years, making it incredibly difficult to completely eradicate.
While significant progress has been made in treatment, shortening regimens from 24 months to the current standard of 4-6 months, this still requires a demanding combination of at least four different antibiotics. The length and difficulty of this regimen often leads patients to end treatment early, which not only allow the bacteria to survive but evolve into multi-drug resistant (MDR) and extensively-drug resistant (XDR) strains of TB.
To overcome these challenges, the ultimate goal is to develop novel, shorter-course drug regimens that can cure TB in less than 8 weeks. A more effective and rapid treatment would drastically improve patient quality of life and prevent the emergence of new resistant strains. This project contributes to this goal by uncovering the specific metabolic vulnerabilities of M. tuberculosis that can be exploited to design the next generation of rational drug combinations.
Project Approach
Under the guidance of Dr. Selva Rupa Christinal Immanuel and Dr. Mario Arrieta-Ortiz in the Baliga Lab, this project took a systems-level approach to ask a critical question: How does M. tuberculosis adapt its metabolism when faced with different nutrient landscapes inside the human body? How can this understanding potentiate drug effectiveness?
The work by Jonathan Chen and Sihan Chen focused on the in silico analysis and interpretation of two large experimental datasets.
The project began with a set of over 10 target genes identified by PRIME, a predictive computational model developed by Drs. Immanuel and Arrieta-Ortiz. PRIME integrates Mtb’s regulatory and metabolic networks to predict “conditional vulnerabilities”—genes that become essential for the bacterium’s survival only under specific environmental conditions. Using CRISPRi technology, these target genes were repressed to create knockdown strains, which then formed the basis of the experimental analysis.
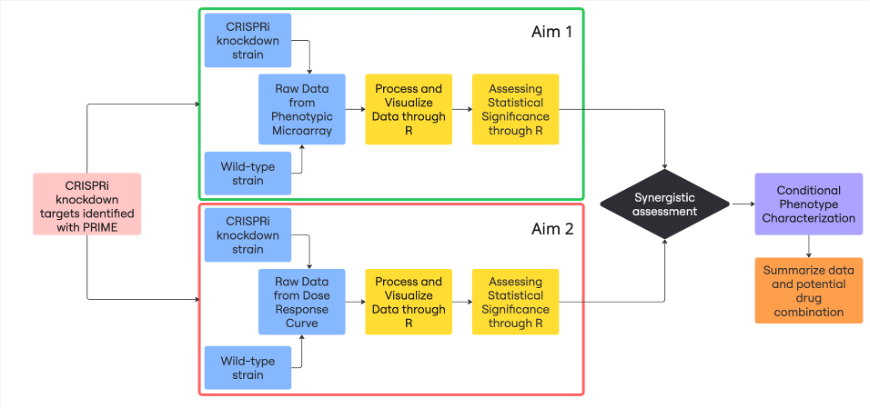
Part 1: Omnilog Phenotypic Microarray Screens
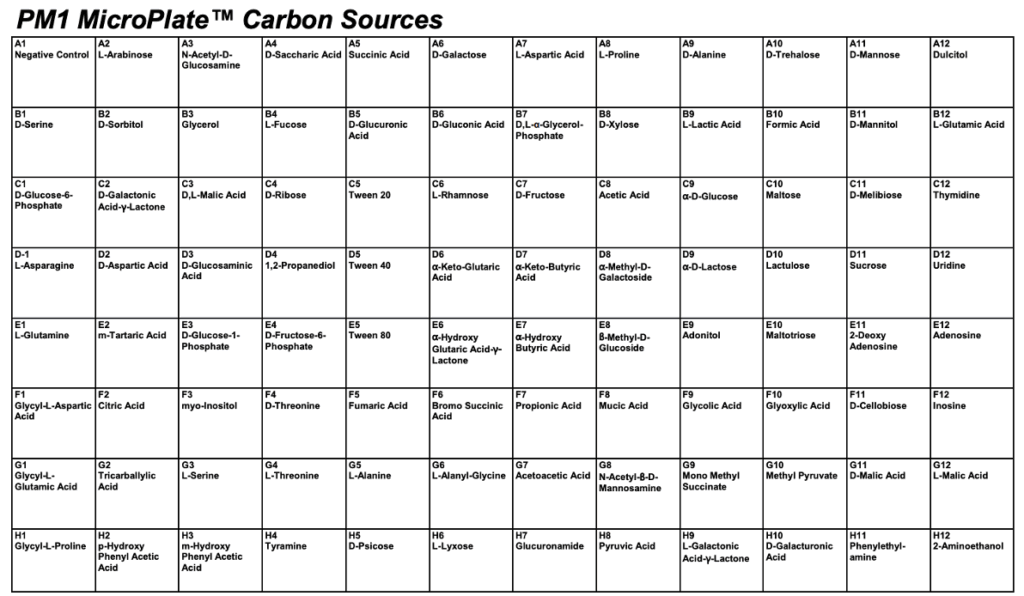
The first phase of the project involved characterizing the metabolic consequences of repressing each target gene. This was achieved using data from high-throughput Omnilog phenotypic microarray screens.
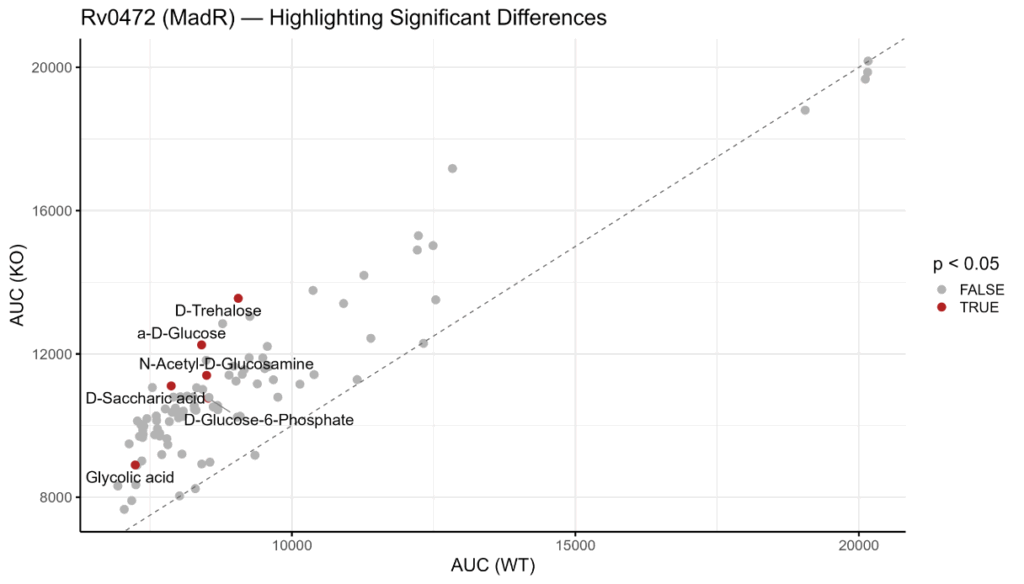
Jonathan and Sihan developed and implemented an R-based computational pipeline to process the raw respiration data from these screens. The pipeline calculated the Area Under the Curve (AUC) to quantify total metabolic activity for each strain on 96 distinct carbon sources. By normalizing this activity against a “no carbon” control and performing statistical tests (t-tests), they identified substrates that were critical for the survival of each knockdown strain.
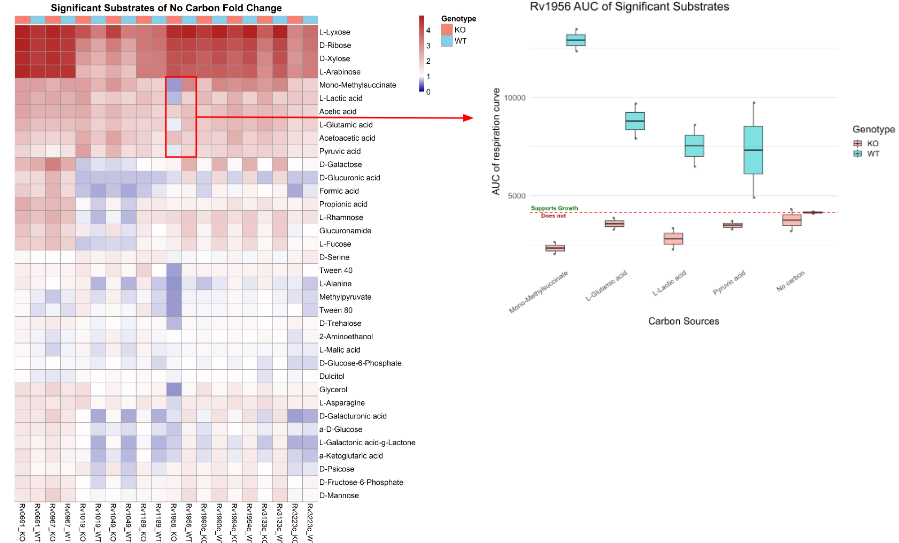
Part 2: Minimum Inhibitory Concentration Data
The second phase integrated these metabolic findings with drug efficacy. The same CRISPRi knockdown strains were treated with a panel of nine different anti-tubercular drugs to generate dose-response curves.
A second R pipeline was developed to analyze the Minimum Inhibitory Concentration (MIC) data from these experiments. By calculating the IC50 value—the drug concentration required to inhibit 50% of bacterial growth—they could quantify how knocking down a specific gene impacted the potency of each drug. This allowed them to identify “sensitizing” knockdowns, where perturbing a metabolic pathway made the bacterium significantly more vulnerable to a specific antibiotic.
Conclusions and Key Takeaways
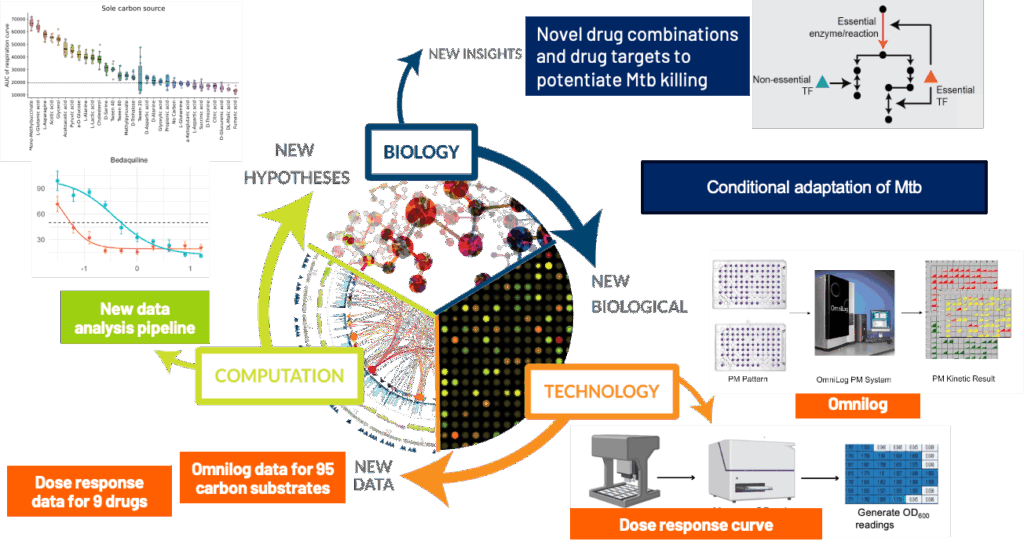
By integrating the analysis from the phenotypic microarray screen and minimum inhibitory concentration data, this work established links between Mtb’s metabolic pathways and its ability to withstand antibiotic stress. The analysis revealed specific genetic perturbations that create vulnerabilities to existing drugs, providing a mechanistic basis for proposing novel, synergistic drug combinations.
CRISPRi and metabolic stress reveals adaptation strategies – By silencing 10 predicted genes and exposing the strains to diverse carbon sources, they uncovered how TB reprograms its metabolism to survive under nutrient stress.
Drug synergy from conditional knockdowns – Testing nine antibiotics on these strains showed that some gene knockdowns potentiate drug activity, revealing new targets for combination therapy.
Systems biology offers predictive power – Their approach shows that combining computational modeling with experimental data can forecast pathogen behavior and identify strategies to outmaneuver it.
This work supports next-gen TB treatment design – By identifying metabolic vulnerabilities and drug-sensitizing targets, they are laying the groundwork for more effective, personalized TB regimens.